Genomic comparison and phenotypic characterization of Pseudomonas aeruginosa isolates across environmental and diverse clinical isolation sites
Abstract
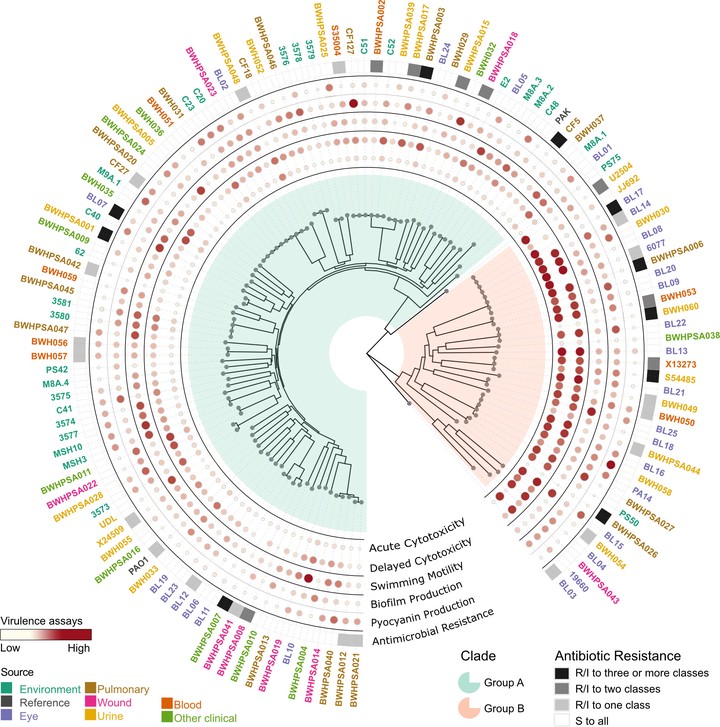
Pseudomonas aeruginosa is a clinically significant, opportunistic pathogen adept at thriving in both host-associated and environmental settings. We sought to define the extent to which P. aeruginosa isolates specialize across niches using a comprehensive study of whole-genome sequencing with paired phenotypic characterization of 125 P. aeruginosa isolates from diverse clinical and environmental sites. We evaluated virulence-associated traits, including motility, cytotoxicity, biofilm formation, pyocyanin production, and antimicrobial resistance to eight antibiotics. Our results show that genomic diversity does not correlate with isolation source or most virulence phenotypes. Instead, we find that, in agreement with prior studies, the two major P. aeruginosa clades (groups A and B) clearly segregate by cytotoxicity, with group B strains showing significantly higher cytotoxicity than group A. Sequence analysis revealed previously uncharacterized alleles of genes encoding type III secretion effector proteins. We observed high variability among strains and isolation sources in the four assayed virulence phenotypes. Antimicrobial resistance was exclusively observed in clinical isolates, whereas it was absent in environmental isolates, reflecting antibiotic exposure-driven selection. Bacterial genome-wide association studies (GWAS) revealed an association between cytotoxicity and exoU presence, and we identified a novel exoU allelic variant with decreased cytotoxicity, demonstrating that functional diversity of well-characterized virulence factors may influence pathogenic outcomes. Overall, our analysis supports the hypothesis that the ability of P. aeruginosa to thrive across diverse niches is driven not by niche-specific accessory genes but by its core genome.
CP and EB are co-primary authors. CP, JR, and DH are co-corresponding authors.